








Sticklerov syndróm
Hereditárna artro-oftalmopatia
Popis:
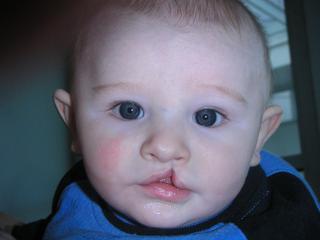
Sticklerov syndróm (ang. Stickler syndrome) je genetická porucha, ktorá spôsobuje vážne poruchy zraku, sluchu, pohybového aparátu a ďalšie problémy. Je tiež známy ako dedičná progresívna ARTHRO-oftalmopatia. Sticklerov syndróm je zvyčajne diagnostikovaný už počas detstva. Deti, ktoré majú Sticklerov syndróm majú často charakteristické rysy tváre: sploštená tvár, výrazné oči, malý nos, chudý vzhľad tváre, dlhá horná pera (philtrum), a malá vystupujúca brada. Často sa rodia s rázštepom podnebia. Príznaky syndrómu sa však môžu značne líšiť od človeka k človeku.
Diagnostika:
Vzhľadom k typickým abnormalitám v tvári je diagnóza často odhalená už krátko po pôrode. Je ale veľa prípadov, kedy bol tento syndróm diagnostikovaný až oveľa neskôr v detstve, niekedy aj v čase dospievania. Pre stanovenie diagnózy je nutné oftalmologické (očné), ORL (ušné, nosné, krčné), reumatologické, ortopedické a röntgenologické vyšetrenie (Rtg). Na základe ich posudku sa odporúča genetické vyšetrenie (najčastejšie odber krvi) a následne starostlivosť genetika.Sticklerov syndróm postihuje odhadom 1 z 7500 až 9000 novorodencov. Odborníci popísali niekoľko typov tohto syndrómu, ktoré sa líšia podľa príčiny a ich charakteristických príznakov a symptómov. Najmä očné abnormality a závažnosť straty sluchu sa líši medzi jednotlivými typmi syndrómu.
Sticklerov syndróm typu I (STL1): je zodpovedný za približne 70% hlásených prípadov a predstavuje veľmi širokú škálu príznakov, ktoré sa týkajú ako očí, uší, vzhľadu tváre, poschodia a pohybového aparátu. Dochádza k nemu v dôsledku mutácií celého COL2A1 génu na chromozóme 12q13 0,11. Tieto mutácie spôsobujú stratu funkcie COL2A1 génu. Táto mutácia spôsobuje malý vzrast a skorý nástup osteoartrózy. Dedičnosť tohto typu je autozomálne dominantné.
Sticklerov syndróm typu II (STL2): dochádza k nemu v dôsledku mutácií COL11A1 génu na chromozóme 1p21. Pacienti s iným, dosť podobným ochorením, tzv. Marshallovým syndrómom, môžu mať tiež mutácie COL11A1 génu, ale pacienti sa Sticklerovým syndrómom typu II majú miernejší fenotyp s menej výraznými črtmi tváre ako pacienti s Marshallovým syndrómom. Ľudia s STL2 majú menej výrazné sploštenie tváre a nosový mostík je rozvinutejší ako u pacientov s Marshallovým syndrómom. Krátkozrakosť a retinálna degenerácia nemusí byť vždy prítomná. Sivý zákal a vážnejšie skorý nástup straty sluchu je však u tohto syndrómu častejší. Dedičnosť Sticklerovho syndrómu typu II je tiež autozomálne dominantná.
Sticklerov syndróm typu III (STL3): bol opísaný ako forma bez očných problémov. U tohto typu nebývajú ani krátkozrakosť, ani zákaly, ale je tu väčšia pravdepodobnosť postihnutia kĺbov. STL3 je spôsobený mutáciami COL11A2 génu na chromozóme 6p21.3. Dedičnosť je tiež autozomálne dominantná. Táto forma je vyvolaná rovnakým typom mutácie a podobnými príznakmi ako heterozygotná oto-spondylo-megaepiphyseal dysplázia (OSMED), s ktorým sa toto ochorenie môže ľahko zameniť.
Sticklerov syndróm typu IV (STL4): je spôsobený mutáciou v géne COL9A1, ktorý sa nachádza na chromozóme 6q13. Tento typ bol zistený a nahlásený v Turecku a Maroku u príbuzného klanu rodiny. Dedičný vzor bol autozomálne recesívny (postihnutý gén majú obaja rodičia, ale u nich sa choroba neprejavila).
Sticklerov syndróm typu V (STL5): je pravdepodobne spôsobený mutáciou COL9A2 génu, ktorý sa nachádza na chromozóme 1p33. Tento typ bol zachytený iba v rodine v Indii. Dedičný vzor je rovnako ako u typu IV autozomálne recesívny. U tohto typu syndrómu bola veľmi nedávno zistená tiež mutácie génu COL9A3. Boli nahlásené tri prípady bratov v marockej rodine, ktoré majú rovnaké príznaky + mentálne postihnutie.
Sticklerov syndróm je spôsobený mutáciami v génoch podieľajúcich sa na tvorbe kolagénu (jedna zo stavebných látok mnohých typov spojivových tkanív). Dedičný je autozomálne dominantný dedičným spôsobom. To znamená, že jeden z rodičov musí mať dominantný (nadradený) postihnutý gén. Pokiaľ ho má, je sám postihnutý a je 50% pravdepodobnosť, že ochorie aj niektorý z jeho potomkov. Zriedkavo však môže dôjsť k mutácii génu úplne náhodne (primárne).
Liečba:
Tak ako u všetkých genetických porúch, ani pre túto genetickú poruchu neexistuje žiadny liek. Liečba sa zameriava na známky a príznaky ochorenia (je symptomatická). Analgetiká môžu zmierniť opuchy, bolesti a stuhnutosť kĺbov. Glaukóm v podobe očných kvapiek reguluje tlak vo vnútri očí. Pri poruchách sluchu sa používajú slúchadla alebo aj kochleárny implantát. Pri poruchách sluchu je nutné pomáhať deťom s vývojom reči (logopéd). A veľmi dôležitá je aj fyzikálna terapia. Rôzne podpery, palice, cviky atď. môžu pomôcť zlepšiť stuhnutosť kĺbov. Niekedy je nutná aj chirurgická liečba (rázštepy pier, tracheostomia u detí, ktoré majú problém s dýchaním, predĺženie spodnej čeľuste, preťahovanie kostí atď.). U častých ušných infekciách sa robí drenáž stredného ucha, vykonávajú sa operácie očných zákalov a iné. Tiež je často potrebný špeciálny pedagóg (ťažkosti s učením v normálnej škole). Pravidelné návštevy u odborných lekárov sú celoživotnou nutnosťou. Ochorenie je totiž nutné neustále sledovať a na každé zhoršenie problémov rýchlo reagovať. Napríklad sluch by mal byť kontrolovaný každých 6 mesiacov do veku 5 rokov dieťaťa a potom aspoň raz ročne. Podobné je to aj s očným vyšetrením.
Prejavy:
- Očné problémy: predovšetkým ťažká krátkozrakosť, šedý zákal a glaukóm (zelený zákal) a odlúčenie sietnice. Postihnutí jedinci môžu mať tenkú vrstvu nervových očných buniek v sietnici, ktorá lemuje zadnú časť oka (vitreoretinálna degenerácia). Sietnica sníma svetlo a prevádza ho na nervové signály, ktoré sú následne odovzdané mozgu prostredníctvom očného nervu. Vitreoretinálna degenerácia môže spôsobiť drobné škvrny (vločky), ktoré zmenšujú zorné pole a tým zhoršujú videnie. Okrem toho môže tiež spôsobiť odlúčenie sietnice (sietnice sa oddelí od základneho tkaniva). V niektorých prípadoch môže dôjsť k trhlinkám v sietnici. Ľudia majú spočiatku rozmazané videnie, náhle záblesky svetla a náhle zhoršenie zraku. Je to akoby mal človek cez oči závoj. Ak sa nezačne včas s liečbou, postupne dôjde k úplnej strate zraku. Odlúčenie sietnice sa môže objaviť v každom veku. Ďalšími očnými problémy tohto syndrómu sú: zakalenia (nepriehľadnosť) očných šošoviek (katarakty), skríženie očného videnia (strabizmus) a abnormálne zakrivenie rohovky. U 5-10% ľudí sa postupne vyvinie zvýšený tlak v oku, ktorý spôsobuje charakteristické poškodenie očného nervu.
- Sluchové problémy: rozsah straty sluchu je u každého postihnutého iný, ale takmer vždy postihuje schopnosť počuť vysoké frekvencie. K strate sluchu dochádza v dôsledku ako prevodnej, tak i percepčnej nedoslýchavosti. Jednak bývajú problémy stredného ucha (predovšetkým zlá funkcia sluchových kostičiek) alebo neschopnosť sluchového nervu viesť vzruchy do mozgu. Strata sluchu môže byť jednostranná aj obojstranná, čiastočná aj úplná. Časté sú infekcie stredného ucha (zápaly) a veľká tvorba ušného mazu.
- Kostí a kĺbové abnormality: postihnutí majú často príliš pružné kĺby a majú veľkú pravdepodobnosť vzniku abnormálneho zakrivenia chrbtice, ako je napríklad skolióza, kyfóza a iné. Artróza môže začať už v období dospievania. Mnoho jedincov má problémy so zápalmi kĺbov v priebehu tretej a štvrtej dekády života (osteoartritída). Môžu nastať rôzne deformity hrudníka, ako je napr. Vpáčený hrudník (stlačenie hrudnej kosti). Ďalej môžu byť nezvyčajne dlhé, štíhle prsty (arachnodactyly), ploché nohy (PES planus), a deformácie bokov a veľa ďalších.
- Dýchacie ťažkosti a spôsob prijímania potravy: Dýchanie alebo prijímanie potravy spôsobuje ťažkosti nielen deťom s rázštepom pery, ale aj u detí s malou spodnou čeľusťou (mikrognacia) a s problémami s jazykom (tendencia neovládať dobre jazyk, ktorý padá smerom k hrdlu, čo je spôsobené jeho umiestnením, ktoré je viac vzadu než je normálny jazyk (glossoptosis)
- Srdcové problémy: niektorí ľudia môžu mať vyššie riziko problémov so srdcových chlopní.
- Zubné problémy: väčšina detí, ktoré majú Sticklerov syndróm, majú abnormálne malú čeľusť, preto často nie je dosť miesta pre rast plného počtu dospelých zubov.
Na mape sú vyznačené rodiny so syndrómom - Sticklerov syndróm
Po registrácii môžete tieto rodiny kontaktovať. Prípadne Vás v budúcnosti môže kontaktovať druhá rodina, s ktorou si môžete vymeniť informácie.
Ak máte tento syndróm, zaregistrujte sa