








Ataxia Telangiektázia
AT syndróm, Louis-Barovej syndróm, Cerebello-okulokutánna tellangiektázia
Ataxia telangiectázia (AT) je komplexná genetická neurodegeneratívna porucha, ktorá sa môže prejaviť v detstve alebo ranom detstve. Porucha je charakterizovaná progresívne zhoršenou koordináciou dobrovoľných pohybov (ataxia), rozvojom červenkastých lézií na koži a slizniciach v dôsledku trvalého rozširovania skupín krvných ciev (telangiektázia) a zhoršenou funkciou imunitného systému (tj. a humorálnej imunodeficiencie), čo má za následok zvýšenú náchylnosť na infekcie horných a dolných dýchacích ciest (sinopulmonálne infekcie). Jedinci s AT majú tiež zvýšené riziko vzniku niektorých malignít, najmä lymfatického systému (lymfómy), krvotvorných orgánov (napr. Leukémie) a mozgu.
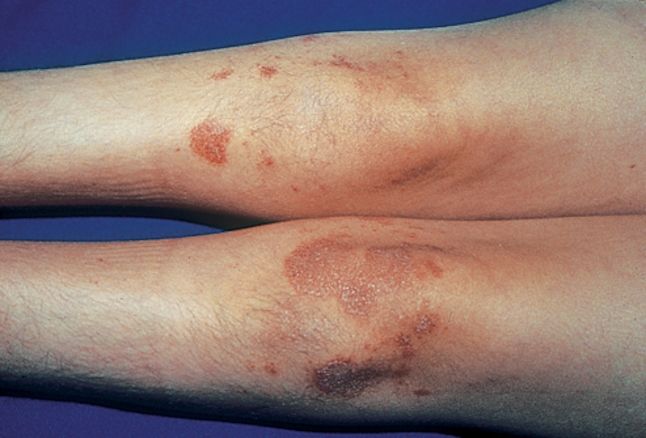
U pacientov s AT sa progresívna ataxia typicky vyvíja v detstve a na začiatku môže byť charakterizovaná abnormálnym kývaním hlavy a trupu. Ako choroba postupuje, stav vedie k neschopnosti chodiť (ambulácia) v neskorom detstve alebo dospievaní. Ataxiu často sprevádzajú ťažkosti s rozprávaním (dyzartria), slintanie; a zhoršená schopnosť koordinovať určité pohyby očí (okulomotorická apraxia), vrátane výskytu nedobrovoľných, rýchlych, rytmických pohybov (oscilácií) očí pri pokuse zamerať sa na určité objekty (fixačný nystagmus). Postihnuté deti môžu tiež vyvinúť neobvykle sklonený postoj a nepravidelné, rýchle, trhavé pohyby, ktoré sa môžu vyskytnúť v spojení s relatívne pomalými, zvíjavými pohybmi (choreoatetóza). Do polovice detstva sa navyše môže vyvinúť teleangiektázia, ktorá sa často objavuje na oblastiach pokožky vystavených slnku, ako je nosový mostík, uši a určité oblasti končatín, ako aj na očných slizniciach ( spojivka).
AT je dedičná ako autozomálne recesívny znak. Porucha je spôsobená zmenami (mutáciami) génu známeho ako ATM (pre „mutované AT“), ktorý bol mapovaný do dlhého ramena (q) chromozómu 11 (11q22.3). Gén ATM riadi (kóduje) produkciu enzýmu, ktorý hrá úlohu v regulácii delenia buniek po poškodení DNA.
Diagnostika:
Ataxia telangiektázia je dedičná ako autozomálne recesívny znak. Genetické choroby sú určené dvoma génmi, jedným prijatým od otca a druhým od matky.
Recesívne genetické poruchy sa vyskytujú vtedy, ak jedinec zdedí rovnaký abnormálny gén pre rovnakú vlastnosť od každého rodiča. Ak jednotlivec dostane jeden normálny gén a jeden gén na ochorenie, bude osobou nosičom choroby, ale spravidla nebude vykazovať žiadne príznaky. Riziko, že dvaja rodičia - nositelia prechádzajú defektným génom, a teda majú postihnuté dieťa, je 25% pri každom tehotenstve. Riziko mať dieťa, ktoré je nosičom ako rodičia, je pri každom tehotenstve 50%. Šanca dieťaťa získať normálne gény od oboch rodičov a byť geneticky normálna pre tento konkrétny znak je 25%.
Gén choroby, ktorý spôsobuje ataxiu telangiektáziu, známy ako gén ATM, sa nachádza na dlhom ramene (q) chromozómu 11 (11q22.3). Chromozómy sa nachádzajú v jadre všetkých telových buniek. Nesú genetické vlastnosti každého jednotlivca. Páry ľudských chromozómov sú očíslované od 1 do 22, s nerovnakým 23. párom chromozómov X a Y pre mužov a dvoma chromozómami X pre ženy. Každý chromozóm má krátke rameno označené ako „p“ a dlhé rameno označené písmenom „q“. Chromozómy sa ďalej delia na pásy, ktoré sú očíslované.
Vedci zistili, že gén ATM kóduje proteín, ktorý hrá úlohu v regulácii delenia buniek po poškodení DNA (DNA alebo deoxyribonukleová kyselina je nosičom genetického kódu). Proteín, ktorý je známy ako ATM pre „mutované AT“, je enzým (proteínkináza), ktorý normálne reaguje na poškodenie DNA spustením akumulácie proteínu (p53 ), ktorá bráni deleniu buniek (tumor supresorový proteín). U jedincov s ataxia telangiektáziou však abnormálne zmeny (mutácie) ATM génu spôsobujú absenciu alebo defekt ATM proteínu a oneskorenú akumuláciu proteínu p53. Bunky s poškodením DNA sa v dôsledku toho naďalej delia (replikujú) bez vhodnej opravy svojej DNA, čo spôsobuje zvýšené riziko vzniku rakoviny. Predpokladá sa, že približne polovica ľudských rakovín je charakterizovaná abnormalitami ovplyvňujúcimi aktivitu proteínového supresorového proteínu p53. Vystavenie ionizujúcemu žiareniu (ako napríklad röntgenovému žiareniu) normálne zvyšuje aktivitu ATM proteínu smerovanú p53; avšak u jedincov s ataxia telangectasia má deficitná aktivita ATM proteínu za následok extrémnu citlivosť na takéto žiarenie.
Ataxia telangiektázia sa zvyčajne začína v detstve (vo veku od jedného do troch rokov) a často postihuje viac ako jedno dieťa v rodine. Muži a ženy môžu byť postihnutí v rovnakom počte. V USA je prevalencia približne jedna zo 40 000-100 000 živonarodených detí.
Diagnóza ataxie telangiektázie sa stanovuje na základe podrobnej anamnézy pacienta, dôkladného klinického hodnotenia, identifikácie charakteristických symptómov a radu špecializovaných testov vrátane krvných testov, zobrazovania magnetickou rezonanciou (MRI) a karyotypizácie.
Krvné testy môžu odhaliť zvýšené hladiny sérového alfa-fetoproteínu, ktoré sa vyskytujú v približne 85 percentách prípadov. U nepostihnutých detí však tento proteín môže zostať zvýšený až do veku 2 rokov. Krvné testy môžu tiež odhaliť zvýšené pečeňové enzýmy. Počas MRI sa magnetické pole a rádiové vlny používajú na vytváranie prierezových obrazov mozgu, ktoré môžu vykazovať progresívnu cerebelárnu atrofiu. Karyotypizácia je špecializovaný test, ktorý zisťuje chromozomálne abnormality. Postihnutí jedinci majú zvýšenú frekvenciu takýchto chromozomálnych abnormalít.
Terapia:
Liečba AT je zameraná na kontrolu symptómov. Pri respiračných infekciách môže byť v niektorých prípadoch účinná terapia antibiotikami, posturálna drenáž (s hlavou nižšie ako zvyšok tela) priedušiek a pľúc a injekcie gamaglobulínu.
Vyhýbanie sa neprimeranému slnečnému žiareniu môže pomôcť kontrolovať šírenie a závažnosť rozšírených ciev (telangiektázie). Terapia vitamínom E v niektorých prípadoch bola hlásená ako dočasná úľava od niektorých symptómov, ale mala by byť vyskúšaná iba pod dohľadom lekára a pod dohľadom lekára, aby sa zabránilo toxicite. Liek Diazepam (Valium) môže byť v niektorých prípadoch užitočný na pomoc pri nezrozumiteľnej reči a mimovoľných svalových kontrakciách. Fyzická terapia môže pomôcť udržať svalovú silu a zabrániť kontrakcii končatín. Je potrebné dbať na ochranu pred infekciami.
Ďalšia liečba je symptomatická a podporná. Genetické poradenstvo môže byť prospešné pre osoby s AT a ich rodiny.
Prejavy:
- znížená svalová koordinácia (prejavuje sa väčšinou, keď začína dieťa chodiť)
- koordinácia (najmä v oblasti hlavy a krku) sa zhoršuje a môže dôjsť k chveniu (mimovoľné svalové kontrakcie)
- u väčšiny detí nie je ovplyvnené mentálne fungovanie (väčšina detí prejavuje normálnu alebo nadpriemernú inteligenciu)
- teleangiektázie (viditeľné rozšírené cievy) zvyčajne začínajú v očiach (oči vyzerajú „podliate krvou“) vo veku od troch do šiestich rokov
- možný zvýšený výskyt karcinómu a lymfómu, ktoré sa zvyčajne začínajú v ranej dospelosti
- približne u jedného z troch postihnutých jedincov sa vyvinie rakovina, zvyčajne rakovina niektorých zhubných nádorov, najmä lymfatického systému (lymfómy) alebo krvi (leukémia)
- v niektorých prípadoch sa môže vyskytnúť mierna forma diabetes mellitus (stav, pri ktorom dochádza k nedostatočnej sekrécii hormónu inzulínu)
- k primárnym symptómom môže patriť abnormálne zvýšený smäd a močenie (polydipsia a polyúria), strata hmotnosti, nechutenstvo a únava
Na mape sú vyznačené rodiny so syndrómom - Ataxia Telangiektázia
Po registrácii môžete tieto rodiny kontaktovať. Prípadne Vás v budúcnosti môže kontaktovať druhá rodina, s ktorou si môžete vymeniť informácie.
Ak máte tento syndróm, zaregistrujte sa